
Rumbidzayi Zinyuke
Health Buzz
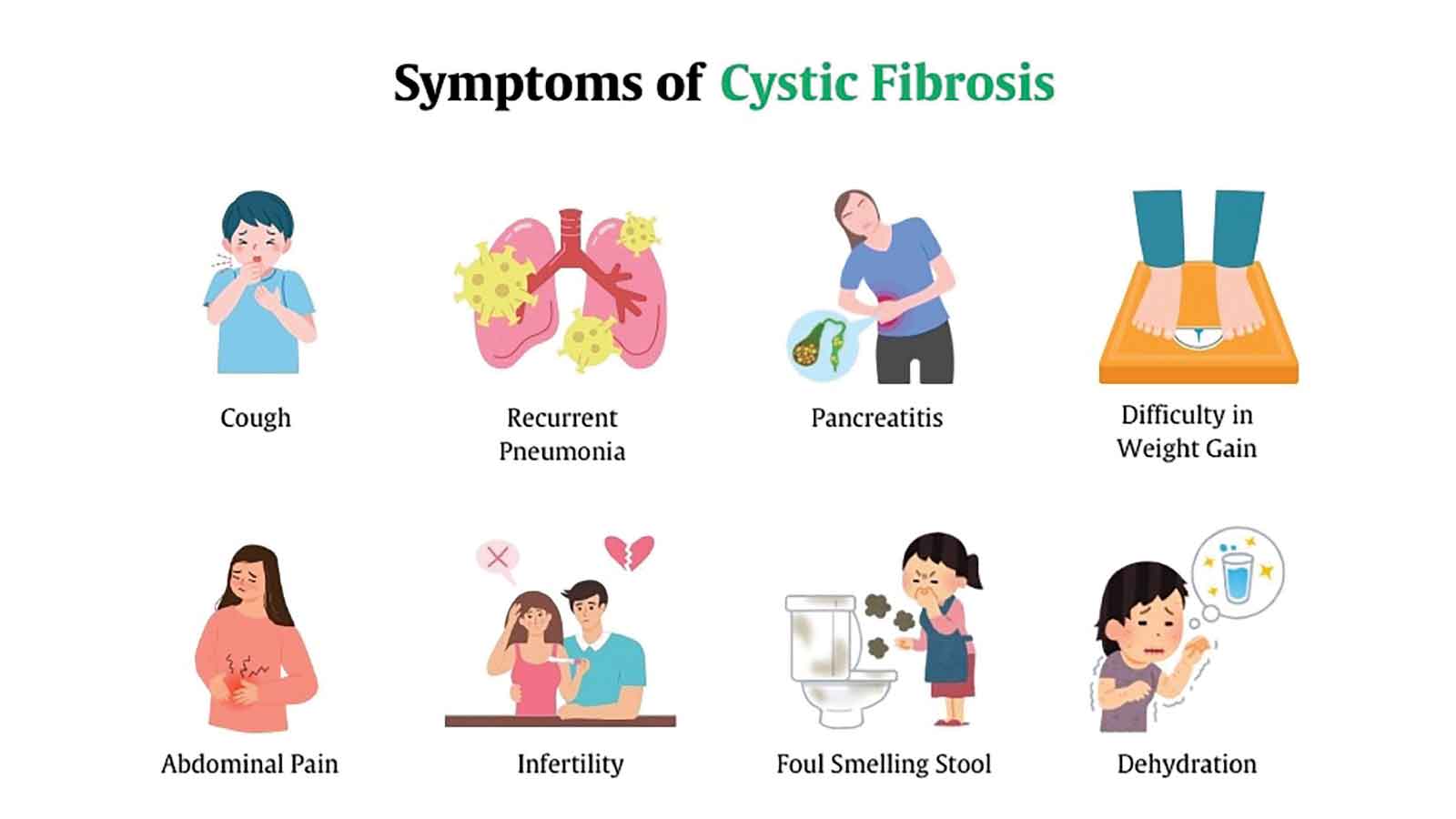
A persistent cough that never quite goes away. Frequent chest infections that keep a child in and out of hospital. A teenager who struggles to gain weight no matter how much they eat.
For many families, these are not isolated health concerns but the daily reality of living with cystic fibrosis, a lifelong genetic condition that quietly affects multiple organs while demanding constant care and vigilance.
Cystic fibrosis (CF) is a hereditary disease that primarily damages the lungs and digestive system. It interferes with the body’s ability to produce normal mucus, sweat and digestive fluids, turning them thick and sticky instead of thin and free-flowing.
These abnormal secretions clog airways and ducts, leading to breathing difficulties, repeated infections and problems with absorbing nutrients essential for growth and development.
Globally, cystic fibrosis is classified as a rare disease, yet its burden is significant. It is estimated to affect about one in every 2 000 to 3 000 newborns in parts of Europe. More than 100 000 people are believed to be living with CF worldwide. Although historically associated with people of European descent, the condition affects all racial and ethnic groups, including populations in Africa where it remains largely underdiagnosed.
In Zimbabwe and across much of sub-Saharan Africa, reliable national statistics on cystic fibrosis are limited. Health experts say this is not because the disease is absent, but because it is often missed.
Many children presenting with symptoms consistent with CF are instead treated for chronic asthma, recurrent pneumonia or malnutrition, conditions that share overlapping clinical features.
This misdiagnosis delays appropriate care, often allowing the disease to progress unchecked.
At the centre of cystic fibrosis is a genetic mutation affecting the CFTR (cystic fibrosis transmembrane conductance regulator) gene. The condition is inherited in an autosomal recessive pattern, meaning a child must receive a defective gene from both parents to develop the disease.
If both parents are carriers, there is a 25 percent chance with each pregnancy that the child will have cystic fibrosis, a 50 percent chance the child will be a carrier, and a 25 percent chance the child will inherit no faulty gene at all. This genetic link underscores one of the most significant risk factors, family history.
However, because carriers often show no symptoms, many families are unaware of their risk until a child is diagnosed. This highlights the importance of genetic awareness and counselling, particularly in settings where routine screening is not available.
The effects of cystic fibrosis are complex and far-reaching. In the lungs, thick mucus blocks the airways and creates an environment where bacteria can thrive. This leads to chronic infections, inflammation and gradual damage to lung tissue. Over time, breathing becomes increasingly difficult, and lung failure remains the leading cause of death among people living with CF.
The digestive system is also heavily affected. The pancreas, which produces enzymes that help break down food, becomes obstructed by thick secretions. As a result, nutrients are not properly absorbed, leading to poor weight gain, vitamin deficiencies and stunted growth in children.
In a country where malnutrition is already a concern, this aspect of CF can easily be overlooked or misinterpreted.
Beyond the lungs and digestive tract, cystic fibrosis can affect the liver, sinuses and reproductive system.
Many males with the condition may be infertile, while some patients develop liver disease due to blocked bile ducts. These wide-ranging effects make CF a complex condition that requires comprehensive, multidisciplinary care.
Symptoms often begin early in life. Infants may present with a persistent cough, wheezing, frequent lung infections and difficulty gaining weight. One of the lesser-known signs is unusually salty skin, which can sometimes be noticed when a parent kisses their child. In some cases, newborns experience intestinal blockage shortly after birth, a condition known as meconium ileus.
Despite its severity, cystic fibrosis is no longer the immediate death sentence it once was. Advances in treatment have significantly improved life expectancy. In high-income countries, many people with CF now live into their 40s, 50s or beyond. This progress has been driven by improved antibiotics, airway clearance techniques, nutritional support and newer therapies that target the underlying genetic defect.
However, these gains are not evenly shared. In many low- and middle-income countries, including Zimbabwe, access to specialised care remains limited. There are few dedicated CF centres, and advanced medications are often unavailable or unaffordable. Without early diagnosis and proper management, outcomes remain poor, with many patients not reaching adulthood.
While cystic fibrosis cannot be prevented in the traditional sense because it is genetic, its impact can be reduced through informed decision-making and early intervention.
Genetic testing can identify carriers of the CFTR mutation, allowing prospective parents to understand their risk. In settings where such services are available, genetic counselling can help families make informed reproductive choices.
Early detection is equally critical. Newborn screening programmes, widely used in developed countries, enable diagnosis within weeks of birth. This allows treatment to begin before significant damage occurs, greatly improving long-term outcomes.
Expanding such screening in Africa would be a significant step towards addressing the hidden burden of CF.
Awareness remains one of the most powerful tools in changing the trajectory of the disease. When healthcare workers and communities are familiar with the signs and symptoms, children are more likely to be referred for appropriate testing.
Recognising patterns such as repeated chest infections, failure to thrive and persistent digestive problems can make the difference between early intervention and delayed diagnosis.
There is also a human side to cystic fibrosis that goes beyond clinical symptoms.
Experts say managing the disease requires daily treatments, including physiotherapy to clear mucus, regular medication and frequent medical reviews. For children and adolescents, this can interfere with schooling and social life, while families often face emotional strain and financial challenges in providing ongoing care.
Globally, research continues to bring hope. New therapies that correct the function of the defective CFTR protein are transforming care in some parts of the world, offering improved quality of life and longer survival.
However, their high cost remains a major barrier, particularly in resource-limited settings.
For Zimbabwe, integrating cystic fibrosis into broader health strategies could help close the gap. Strengthening diagnostic systems, training healthcare professionals and incorporating CF into child health and non-communicable disease programmes would improve detection and management. Partnerships with international organisations could also support access to essential medicines and technologies.
Ultimately, cystic fibrosis is a reminder that rare diseases still matter. Its low visibility does not diminish its impact. For affected families, it is a daily battle, one that requires resilience, support and access to care.
As awareness grows and science advances, there is hope that more children born with cystic fibrosis will not only survive but thrive. Until then, the focus must remain on early detection, education and equitable access to treatment, ensuring that no child’s breath is cut short by a disease that can be managed with the right support.
Feedback: [email protected]

